[ Home Page ] [ Site Map ]
The Chronic Myeloproliferative Disorders
The chronic myeloproliferative disorders are characterised by over-production of one or more myeloid cell-lineages. They result from the proliferation of a neoplastic clone of cells derived from a multipotent haemopoietic stem cell, and the leukaemic clone preserves the ability to differentiate and mature. The myeloproliferative disorders differ from the myelodysplastic syndromes, which are largely characterised by quantitative and functional deficiencies in cell production.
Classification
Chronic Myeloid Leukaemia (CML) is a malignant disorder of the haemopoietic stem cell usually associated with the presence of the Philadelphia chromosome (Ph), a shortened chromosome 22 resulting from a t(9;22) translocation, and at a molecular level by BCR-ABL gene rearrangement. The emergence of the BCR-ABL fusion protein in the stem cell results in the generation of clonal progeny with a selective growth advantage. CML has three recognisable phases; chronic, accelerated, and blastic transformation in which the condition resembles acute leukaemia.
Juvenile CML is a separate clinical condition, not usually associated with the Ph chromosome, and may have trisomy 8, monosomy 7, or be cytogenetically normal. The condition is associated with reversion to foetal haemopoiesis, and does not usually transform to AML.
Clinical Features
- CML occurs at a frequency of 1 - 1.5 per 100 000, is rare below age 20, with a median age 40-50 years, and is slightly more common in males
- 90% present in chronic phase, 10% picked up as 'incidental' finding during blood test for unrelated reasons
- Splenomegaly, weight loss and fatigue due to anaemia are common presenting features; fever and hepatomegaly are less common. About 20% have no symptoms at presentation
- CML rarely presents in accelerated or blast phase - symptoms may include rapidly advancing weight loss, bleeding, sweating, splenic and bone pain, and fever.
Laboratory Diagnosis
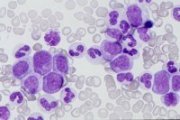
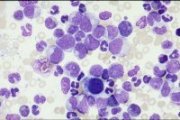
Chronic phase: Peripheral blood leucocytosis, reflected in a numerical increase in all stages of myeloid differentiation. Basophilia frequently pronounced. Bone marrow aspirates usually shows profound hypercellularity and increased myeloid:erythroid ratio (>25:1). Philadelphia chromosome present in all myeloid cells.
 |
 |
|
CML chronic phase: peripheral blood smear |
CML chronic phase: bone marrow aspirate |
CML chronic phase: bone marrow trephine H & E stain (right). Normal reticulin (left). |
Accelerated and blastic phases: Characterised by the emergence of a discrete blast cell population and left-shift compared to chronic phase. Definition of accelerated phase requires at least one of the following:
- rising leucocyte count during adequate treatment
- platelet count <100, or >1000 x 109/l
- haemoglobin <8.0g/dl
- >10% blasts in peripheral blood
- >20% basophils and eosinophils in peripheral blood
Blastic phase is defined as >30% blasts and promyelocytes in peripheral blood or marrow. Normal marrow erythropoiesis and megakaryopoiesis reduced.
|
|
|
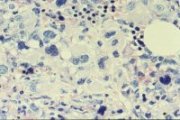
CML blast transformation: myeloid, with increased basophil precursors; peripheral blood |
CML blast transformation: lymphoblastic; peripheral blood |
CML blast transformation: megakaryoblastic; peripheral blood, one micro-megakaryoblast |
 |
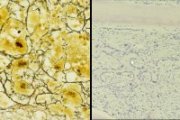 |
CML blast transformation: megakaryoblastic; bone marrow trephine |
CML blast transformation: megakaryoblastic with new bone formation (Giemsa stain, right). Bone marrow reticulin stain (left) |
Morphology and Cytochemistry
The blast cells seen in accelerated and blastic phases of CML do not fit the established FAB criteria. About 70% of cases in blast transformation have a myeloblast phenotype (CD13, CD33, sometimes erythroid or megakaryocyte markers), 20% have a lymphoblast (Tdt, CD10, CD19, CD22), and 10% a mixed phenotype.
Cytogenetics
Demonstration of the Philadelphia chromosome is performed using standard cytogenetic techniques on leukaemic cells in metaphase. The frequency of the Ph chromosome in marrow samples reflects the degree of marrow replacement by the leukaemic clone. FISH techniques can be used to confirm the presence of BCR-ABL on the Ph chromosome 22 (or rarely chromosome 9), and is particularly useful in Ph- cases of CML, and in cases with complex translocations. Southern- and Western-blot analyses can also be used to demonstrate BCR-ABL. RT-PCR is especially useful in the detection of BCR-ABL for analysis of minimal residual disease.
Prognosis and Treatment
- duration of chronic phase 2-6 years, but may be up to 10 or 15 years.
- treatment in chronic phase involves cytotoxic drug therapy, with hydroxyurea and alpha-interferon proving most effective, and busulphan less so.
- leucopheresis is effective in reducing the morbidity due to a high circulating leucocyte mass, and the cells contain a useful source of stem cells which may be cyropreserved and re-infused during later stages of the disease.
- bone marrow transplant is indicated in cases where an HLA-matched close relative is available as donor. A successful transplant in chronic phase gives >50% survival at 5yr, but is rarely successful in accelerated or blastic phases.
- treatment in blastic phase is palliative in nature, as cure is not regarded as a realistic aim, and involves higher doses of hydroxyurea than for chronic phase. Myeloid blast crisis may be treated with anti-AML drugs such as cytosine arabinoside, daunorubicin and etoposide. Duration of myeloid blast crisis rarely exceeds 6 months.
- treatment for lymphoblastic transformation involves anti-ALL drugs such as prednisolone, vincristine, daunorubicin. Remission maybe longer than for myeloid crisis. CNS relapse is commonly seen.

Usually aged 5 or less, predominantly male. The disease is not associated with the Philadelphia chromosome, and produces lower peripheral leucocyte counts than seen in Ph+ CML. There is usually monocytosis and almost always thrombocytopenia. FAB classification of CMML is sometimes recorded. High levels of foetal haemoglobin (Hb-F) are common. 80% of cases lack identifiable genetic abnormalities. Cytotoxic drug treatment is not normally successful, although the disease can be cured by allogeneic, or matched unrelated donor, marrow transplantation.
PRV is an acquired stem cell myeloproliferative disorder, characterised by bone marrow erythroid hyperplasia and increased peripheral blood red-cell volume. Excessive proliferation of the granulocyte and megakaryocyte lines also occurs. PRV has an incidence of 1.8 per 100 000, with an equal male to female ratio.
Symptoms arise from hyperviscocity and vascular damage due to increased red-cell volume. Venous and arterial thromboses are common presenting features, with splenomegaly and hepatomegaly also common.
Laboratory findings usually include raised haemoglobin and packed red-cell volume (haematocrit), but these alone do not constitute PRV. Diagnosis requires the demonstration of a circulating red-cell mass in excess of 36ml/kg, normal arterial O2 saturation, and either splenomegaly, or thrombocytosis (>400 x 109/l) AND leucocytosis (>12 x 109/l). Neutrophils may be 'left-shifted', and absolute basophilia is common. Bone marrow shows erythroid and megakaryocytic hyperplasia, with reduced myeloid:erythroid ratio (<2:1).
|
|
PRV; giant platelet in peripheral blood |
PRV; hypercellular bone marrow trephine |
Median survival in PRV is 9 - 13 years. Treatment includes phlebotomy to reduce the circulating red-cell mass, and hydroxyurea to suppress marrow hyperplasia. 32P and alkylating agents are rarely used now as there is an increased incidence of transformation to acute leukaemia following this type of treatment.
Myelofibrosis consists of several closely related disorders having several features in common, including progressive marrow fibrosis and extra-medullary haemopoiesis. It results from a proliferation of the fibrous connective tissue which supports haemopoietic elements in the bone marrow. It is believed that the fibroblasts are not abnormal, but may be responding to increased growth factors released from abnormal megakaryocytes.
Myelofibrosis occurs at a frequency about 0.5 per 100 000, and is more common in males than females. Peak age at diagnosis is 60 - 70 years. Splenomegaly is common at presentation, sometimes extensive, resulting from splenic haemopoiesis.
Laboratory diagnosis involves finding evidence of extramedullary haemopoiesis including circulating immature myeloid and erythroid cells, tear-drop poikilocytes (abnormal shaped red-blood cells), and increased bone marrow reticulin fibres without known cause. Giant and bizarrely shaped platelet forms may also occur. Mild leucocytosis with a 'left-shifted' granulocyte series, and mild anaemia due to ineffective erythropoiesis are common at presentation.
|
|
Myelofibrosis; peripheral blood showing tear-drop poikilocytes, basophilic stippling and a myelocyte |
Myelofibrosis; bone marrow trephine H & E stain (left). Reticulin stain (right) |
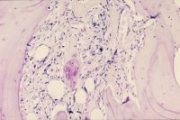 |
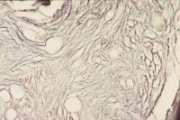 |
Osteomyelosclerosis bone marrow trephine |
Osteomyelosclerosis bone marrow reticulin stain |
Median survival is about 5 years, although it is likely the disease has been progressing for some years before diagnosis. Poor prognostic factors include severe anaemia, thrombocytopenia and the presence of >10% circulating immature myeloid cells. Mortality is usually due to haemorrhagic or thrombotic complications, and transformation to acute leukaemia occurs in about 20% of cases.
ET is characterised by grossly increased peripheral platelet counts, often exceeding 1 x 1012/l. Diagnosis involves the exclusion of other reactive or malignant conditions associated with very high platelet counts, such as a secondary response to chronic blood loss, chronic inflammatory disorders, and other myeloproliferative disorders. Symptoms results from disturbances in platelet function, and include thromboses and haemorrhage. Moderate splenomegaly is also common.
Laboratory findings include dysmegakaryopoiesis, with giant and bizarrely shaped platelets in the peripheral blood. In contrast to other myeloproliferative disorders, basophilia is not usually present. Bone marrow samples are required to demonstrate marked megakaryocyte hyperplasia, and to investigate the presence of the Ph chromosome, fibrosis, and iron deposits for differentiation from other myeloproliferative disorders.
|
|
Essential thrombocythaemia; peripheral blood showing platelet anisocytosis |
Essential thrombocythaemia; bone marrow trephine showing clustered megakaryocytes |
ET is often an indolent condition, with a long median survival of 12 to 15 years. As ET normally occurs in later years, life expectancy is nearly normal at diagnosis. Treatment normally involves managing the sporadic thromboembolic or haemorrhagic episodes. Transition to acute leukaemia occurs less frequently than in other myeloproliferative disorders.
[Home Page ] [Site
Map ]
Copyright © HMDS 1999-2005
Document last updated:
Tuesday, 18 November 2003
Comments & feedback to:
[email protected]
[URL: www.hmds.org.uk/mpd.html]