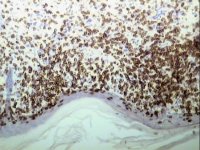
Demonstration of CD2 by streptavidin/ABC on a frozen section of skin
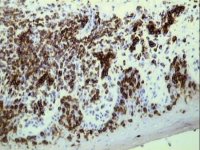
Demonstration of CD5 by streptavidin/ABC on a frozen section of skin
Frozen sections have two main applications in the diagnosis of lymphoproliferative disorders. Firstly in providing a rapid provisional diagnosis and secondly in the demonstration of antigens / enzymes which maybe lost during subsequent fixation and processing schedules.
The freezing procedure is extremely important, if carried out too slowly it will result in the formation of ice crystal artefact, which may make histological interpretation very difficult. For optimum results tissue needs to be sliced thinly (maximum thickness 3mm) and rapidly frozen in liquid nitrogen. Frozen sections are cut at a thickness of 5µm using a cryostat. The sections are dried overnight at 37oC before fixation and subsequent staining or storage.
For subsequent immunocytochemical staining the sections are fixed in acetone for 20 minutes. It is important to dry the sections thoroughly, in order to optimise tissue morphology by reducing the effects of chromatolysis and membrane breakdown which is particularly evident with techniques involving long incubations in aqueous solutions.
|
|
Demonstration of CD2 by streptavidin/ABC on a frozen section of skin |
Demonstration of CD5 by streptavidin/ABC on a frozen section of skin |
Drying the sections overnight at 37oC does not affect section antigenicity. Frozen tissue can be stored in liquid nitrogen or at -70oC for many years without affecting antigenicity. The tissue should be wrapped in aluminium foil or immersed in embedding medium to prevent the tissue from drying out. Similarly, frozen sections can be wrapped in aluminium foil and stored at -20oC for a year or more without loss of antigenicity.
However, no fixative meets all of the above criteria and some compromise is necessary. The choice of fixative will depend upon the type of specimen and the components to be demonstrated.
The most widely used fixatives in diagnostic histology laboratories are formalin based. The three most commonly employed fixatives for general use being neutral buffered formalin, formal saline or as used in HMDS, 10% formalin in tap water.
Formalin, like other aldehyde fixatives, forms cross linking methylene bridges and Schiff bases between basic amino acid (lysine) residues of proteins. This cross linking stabilises the proteins in situ, which is the basis of fixation. Formaldehyde produces mild cross linkages when compared to other aldehyde fixatives such as glutaraldehyde.
In addition to the choice of fixative, other important factors include fixation time, temperature and pH. Fixation time will depend upon the size of the specimen. In order to achieve adequate and consistent fixation it is essential that lymphoreticular specimens be sliced to a maximum thickness of 3mm on arrival in the laboratory. Tissue such as lymph node (3mm slices), skin and bone marrow trephines are routinely fixed for approximately 24 - 48 hours at room temperature. Dense tissue such as spleen may require extended fixation. The rate of fixation can be increased by raising the ambient temperature. However this is not recommended with lymphoid tissue as it has an impaired effect on morphology. The pH of the formalin solution is generally between 5 and 7, which is governed by the pH of the local water supply.
![]()
A haematoxylin and eosin (H&E) stained section is cut from each paraffin block. In addition, a Gordon and Sweet's reticulin stain is performed on all lymph node and spleen cases. After initial examination of the H&E section either additional tinctorial stains or specific panels of immunocytochemical markers are performed.
Before any staining techniques can be employed the methyl methacrylate resin needs to be removed, this is achieved by incubation of the sections in warm (37oC) xylene for 15 minutes and rinsing of the sections in ethanol. We have found that all tinctorial staining techniques which are applicable to paraffin sections will also work on MMA sections. In some instances the staining times may need to be increased. In the case of the H&E the length of time in Harris's haematoxylin is increased to 30 minutes.
 |
 |
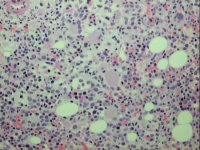
2µm methyl methacrylate resin section of bone marrow trephine biopsy stained with Haematoxylin and Eosin |
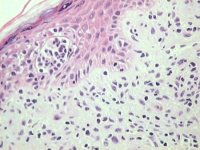
Mycosis fungoides, 2µm MMA section of skin, H & E stained |
After removal of the methyl methacrylate resin as described above, either a conventional Streptavidin-Biotin / Horseradish peroxidase complex technique (StrABC/HRP) or a tyramide amplification of a StrABC/HRP method can be employed. Standard paraffin methodologies can be applied. However, we have found that improved results are obtained by using increased times of antigen retrieval compared to that used for paraffin sections.
 |
 |
 |
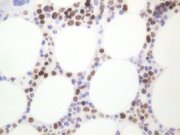
MIB-1 demonstration by streptavidin/ABC method in a MMA section of bone marrow trephine biopsy |
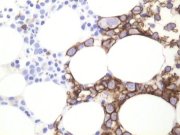
CD20 demonstration by streptavidin/ABC method in a MMA section of bone marrow trephine biopsy |
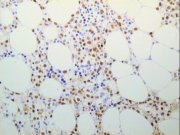
Tdt demonstration by streptavidin/ABC method in a MMA section of bone marrow trephine biopsy |
![]()
Immunocytochemistry is the demonstration of antigens in tissue sections or smears by the use of specific immunological (antibody-antigen) interactions culminating in the attachment of a visible marker to the antigen. The visual marker may be a fluorescent dye, colloidal metal, hapten, radioactive marker or more commonly for light microscopy an enzyme.
Ideally, maximal signal strength along with minimal background or non-specific staining are required to give optimal antigen demonstration.
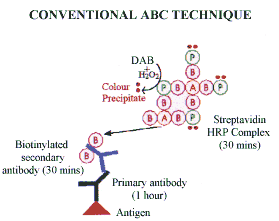
Avidin is a basic glycoprotein (MW 68 Kd) which has a high affinity for the small (MW 24Kd) water soluble vitamin biotin. Biotin can be conjugated to a variety of biological molecules, including antibodies, and many biotin molecules can be attached to a single molecule of protein. The biotinylated protein can thus bind to more than one molecule of avidin.
However, avidin has two distinct disadvantages when used in immunocytochemical detection systems. It has a high isoelectric point of approximately 10 and is therefore positively charged at neutral pH. Consequently it may bind non specifically to negatively charged structures such as the nucleus. The second disadvantage is that avidin is a glycoprotein and reacts with molecules such as lectins via the carbohydrate moiety.
These two problems are overcome with the substitution of streptavidin for avidin. Streptavidin is a protein (MW 60 Kd) isolated from the bacterium Streptomyces avidinii, and like avidin, has four high affinity binding sites for biotin. Streptavidin has an isoelectric point close to neutral pH and therefore possess few strongly charged groups at the near neutral pH used in immunocytochemical detection systems. Furthermore streptavidin is not a glycoprotein and therefore does not bind to lectins. The physical properties of streptavidin therefore make this protein much more desirable for use in immunocytochemical detection systems than avidin.
The most sophisticated and sensitive extension of the streptavidin biotin technique employs pre-formed complexes. Streptavidin and biotinylated horse-radish peroxidase are simply mixed at appropriate concentrations for at least 30 minutes at room temperature for the complex to form. The pre-formed complex is then attached to the biotinylated antibody. Careful stoichiometric control ensures that some binding sites remain free to bind with biotinylated antibody. This allows the pre-formed complex to bind and provides a very high signal at the antigen binding site.
![]()
| The use of biotinylated tyramide to amplify immunocytochemical reactions on tissue sections was first described by Adams et al [2]. The technique requires appropriate pre-treatment procedures and optimal antigen retrieval, and follows a conventional ABC method. After the initial StrABC/HRP layer the biotinylated tyramide step is performed. |  |
|
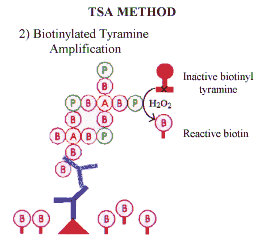 | Horse-radish peroxidase (HRP) in the presence of hydrogen peroxide catalyses the formation of reactive biotinylated tyramide. This reactive biotinylated tyramide covalently binds to proteins via electron rich moieties such as the amino acids tryptophan and tyrosine, resulting in the deposition of biotin at the reaction site. Biotin deposition is restricted to the reaction site since horseradish peroxidase (in the form of StrABC /HRP) is only present at the antigenic site. Subsequent application of labelled streptavidin binds to the newly deposited biotin. | |
| The streptavidin may be labelled with enzyme (eg. HRP) or with appropriate fluorochromes. If enzyme labelled streptavidin is used the reaction can be visualised with the desired chromogen (eg diaminobenzidene). This technique has been used for both immunocytochemistry and for the detection of probes for in situ hybridisation, and represents a significant advance in antigen detection. | 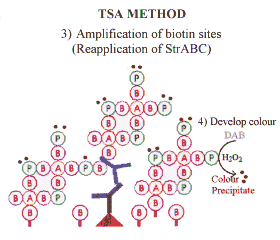 |
|
 |
 |
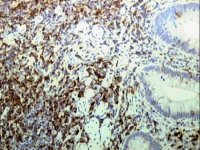
Paraffin section of gastric biopsy showing CD5 positivity by TSA method |
CD2 demonstration by TSA in paraffin section of lymph node from a case of T-cell lymphoma |
 |
 |
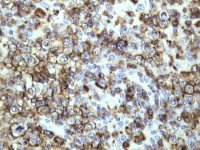
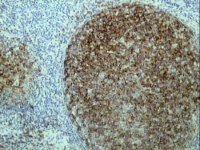
CD10 expression by TSA in paraffin section of lymph node from a case of follicle centre lymphoma |
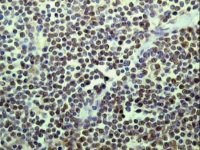
Cyclin D1 demonstration by TSA in paraffin section of lymph node from a case of mantle cell lymphoma |
![]()
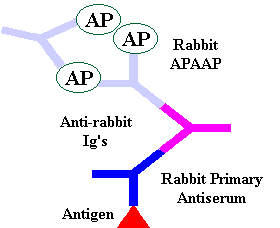 The APAAP technique is an indirect method which utilizes a pre-formed, cyclic enzyme anti-enzyme immuno complex composed of three enzyme molecules (alkaline phosphatase) and two antibody molecules. The technique can be visualised by using either a blue dye (Fast Blue BBN) or a red dye (Fast Red TR). The APAAP technique can be used as an alternative to peroxidase techniques and is particularly useful for double labelling techniques and for antigen demonstration in slides rich in endogenous peroxidase activity, namely peripheral blood and bone marrow aspirate specimens.
The APAAP technique is an indirect method which utilizes a pre-formed, cyclic enzyme anti-enzyme immuno complex composed of three enzyme molecules (alkaline phosphatase) and two antibody molecules. The technique can be visualised by using either a blue dye (Fast Blue BBN) or a red dye (Fast Red TR). The APAAP technique can be used as an alternative to peroxidase techniques and is particularly useful for double labelling techniques and for antigen demonstration in slides rich in endogenous peroxidase activity, namely peripheral blood and bone marrow aspirate specimens.
![]()
The vast majority of antigen retrieval studies have been applied to formalin fixed material. When aldehyde-based fixatives are used (eg. formalin), inter- and intra-molecular cross links are produced with certain structural proteins which are responsible for the masking of tissue antigens. With aldehyde based fixatives this adverse effect has been thought to be due to the formation of methylene bridges between reactive sites on tissue proteins. These reactive sites include primary amines, amide groups, thiols, alcoholic hydroxyl groups and cyclic aromatic rings. The degree of masking of the antigenic sites depends upon the length of time of fixation, temperature, concentration of fixative and the availability of other nearby proteins able to undergo cross-linkages. In order to "unmask" these antigenic sites a range of antigen retrieval techniques are available.
The protein cross links formed during formalin fixation can be partially disrupted by the use of proteolytic enzymes of which trypsin is the most widely used. Trypsinisation time is extremely important and is proportional to the specimen fixation time. There is a very fine balance between over and under digestion. Trypsin is optimally active at 37oC and at pH7.8. The reaction rate is improved by the addition of the co-enzyme calcium chloride (0.1%). Trypsin only remains active for about 30 minutes, therefore if the incubation time exceeds this, the working solution must be replaced. Not all antigens require proteolytic digestion. Furthermore, care must be taken to avoid creating "false" antigenic sites, as some antigens may be altered or destroyed by trypsinisation. In some instances immunostaining may be impaired or completely removed following trypsinisation. Proteolytic digestion has largely been replaced by heat mediated antigen retrieval methods.
The rationale behind these heat pretreatment methods is unclear and several theories have been postulated. One theory is that heavy metal salts (such as the heavy metal solutions described by Shi. et al. [3] act as a protein precipitant, forming insoluble complexes with polypeptides and that protein precipitating fixatives frequently display better preservation of antigens than do cross-linking aldehyde fixatives. Another theory is that during formalin fixation inter- and intra-molecular cross linkages are formed by methylene bridges and weak Schiff bases. These cross linkages alter the protein conformation of the antigen such that it may not be recognised by a specific antibody. It is postulated that heat mediated antigen retrieval removes the weaker Schiff bases but does not affect the methylene bridges so that the resulting protein conformation is intermediate between fixed and unfixed.
Antigens masked during routine fixation and processing can be revealed by using one of the following heat mediated antigen retrieval techniques; microwave oven irradiation, combined microwave oven irradiation and proteolytic enzyme digestion, pressure cooker heating, autoclave heating, water bath heating, Steamer heating, or high temperature incubator
In HMDS we prefer to use microwave irradiation and a combined trypsinisation and microwave irradiation method.
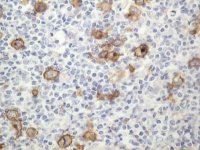 |
Demonstration of CD30 expression by Hodgkin Reed-Sternberg cells in paraffin section of lymph node using streptavidin/ABC method. Optimum results achieved using a combined trypsin and microwave antigen retrieval method. |
![]()
Specimen processing is carried out manually using small glass vials on a roller mixer.
*Antibodies requiring biotinylated tyramide amplification give better demonstration if Vector Antigen unmasking fluid (AUF) is used instead of Citrate buffer. The Vector AUF is used at a dilution of 1/100 in distilled water and consists of a mixture of citric acid and EDTA.
Stir gently at room temperature until solution completely dissolved. Filter through 0.45µm syringe filter.
Allow to thaw out. Immediately before use add 4µl of hydrogen peroxide per 1ml of aliquot.
![]()
Comments & feedback to: [email protected]